
药研头条
阿尔兹海默症药物aducanumab将申请上市 Aβ假说机制迎来逆转
近日,渤健(Biogen)和卫材(Eisai)公司联合宣布,在与FDA和外部专家讨论之后,将计划在明年递交治疗阿尔茨海默症(AD)的在研新药aducanumab的生物制剂申请(BLA)。
今年3月21日,独立数据监督委员会曾对aducanumab出具了无效性分析,认为其达到预期效果可能性很低。因此渤健和卫材终止了aducanumab两项代号为ENGAGE和EMERGE的临床试验。然而,在宣布终止临床试验后,渤健对包含更多患者的数据集进行了分析,发现在EMERGE的临床试验中,剂量为10 mg/kg的aducanumab能够显著改善患者认知能力。同时在名为ENGAGE的临床试验中,持续接受剂量为10 mg/kg的aducanumab的部分患者的认知能力也得到了提高。这一发现让渤健和卫材决定重新启动对aducanumab的申请程序。
Aducanumab临床研究的最新反转无疑为与“淀粉样蛋白(Aβ)假说”的争议性讨论又加了新的话题。近年,多款靶向淀粉样蛋白的在研疗法在III期临床试验中纷纷失败,让人对淀粉样蛋白假说的可信度产生了怀疑。Aducanumab的成功是否意味着“Aβ假说”即将复兴?最近在顶级科学期刊《细胞》上发表的一篇综述表示,虽然现有证据支持淀粉样蛋白在AD病理发生中起到重要的作用,但是淀粉样蛋白引发AD发病的机理可能没有预先想象的那么直接,这仍然是研究人员们需要澄清的问题。
百时美施贵宝取代罗氏成为肿瘤领域霸主
EvaluatePharma近日发布的一份报告显示,罗氏公司在更新抗体专营权、战略交易、临床试验等诸多方面遭遇挫折后,将其在肿瘤学霸主的宝座拱手出让。目前,百时美施贵宝(BMS)对新基的并购最终完成后,将一跃成为全球最大肿瘤学公司,默沙东和阿斯利康预计在未来几年也将强劲增长。
EvaluatePharma预测,在完成并购新基后,从2020年起,至少到2024年,BMS的肿瘤学销售额将超过罗氏,推动这一变化的是Opdivo以及收购新基后获得的重磅免疫调节剂Revlimid。在Revlimid仿制药全面上市前,BMS在2021-2023年期间肿瘤学药物销售额每年将超300亿美元,这将是第一次有公司能打破300亿美元壁垒。
根据EvaluatePharma发布的市场份额预测,在未来几年,肿瘤学市场碎片化将进一步加剧。考虑到医药行业对肿瘤学的持续关注,未来该领域将不断涌入越来越多的小规模参与者。不过,在接下来的几年中,交易、临床挫折和其他意外情况势必会对这一局面带来改变。但唯一可以确定的一点是,肿瘤学市场注定不会平静。
国内药闻 TOP3

2019年《财富》全球未来50强揭晓,恒瑞医药位列第14位
近日,《财富》杂志发布了全球未来50强公司榜单,共有16家中国企业上榜,恒瑞医药位列第14位,是唯一入选的中国制药企业。
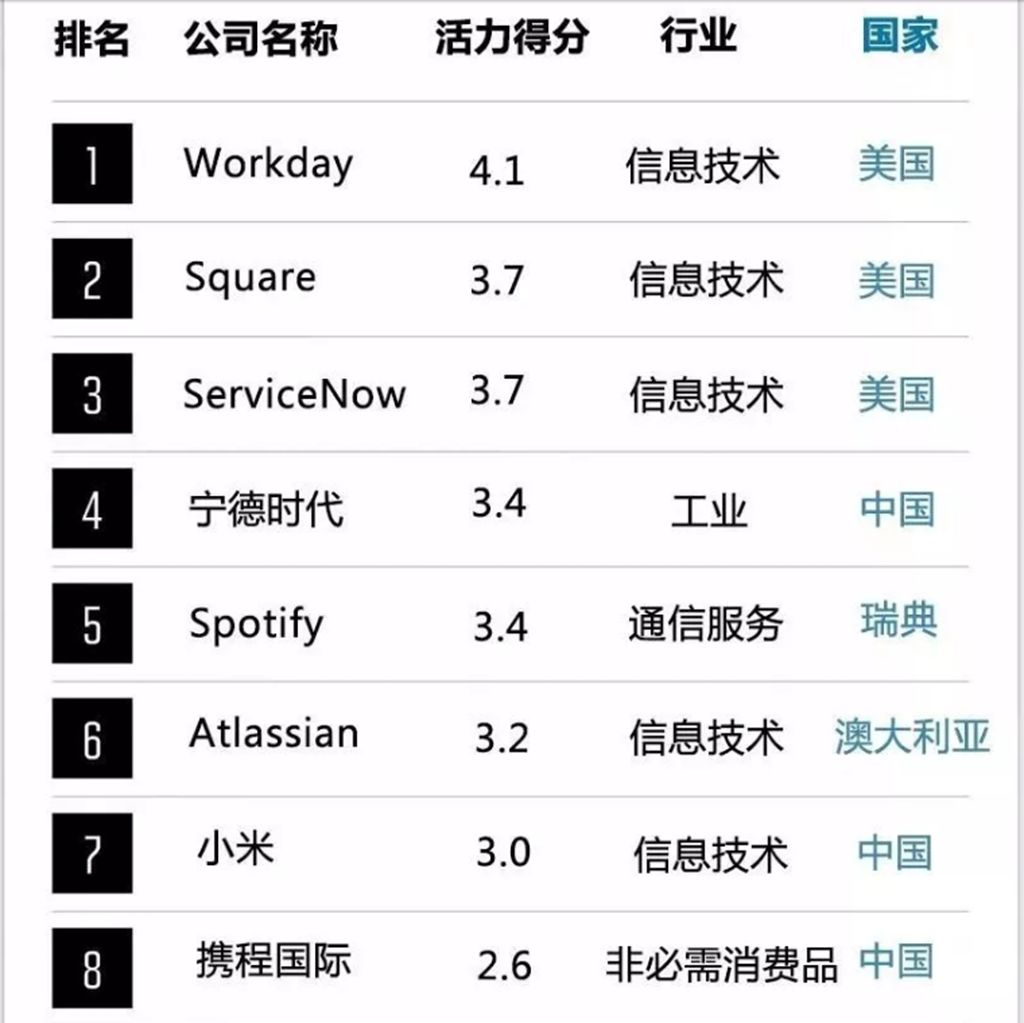
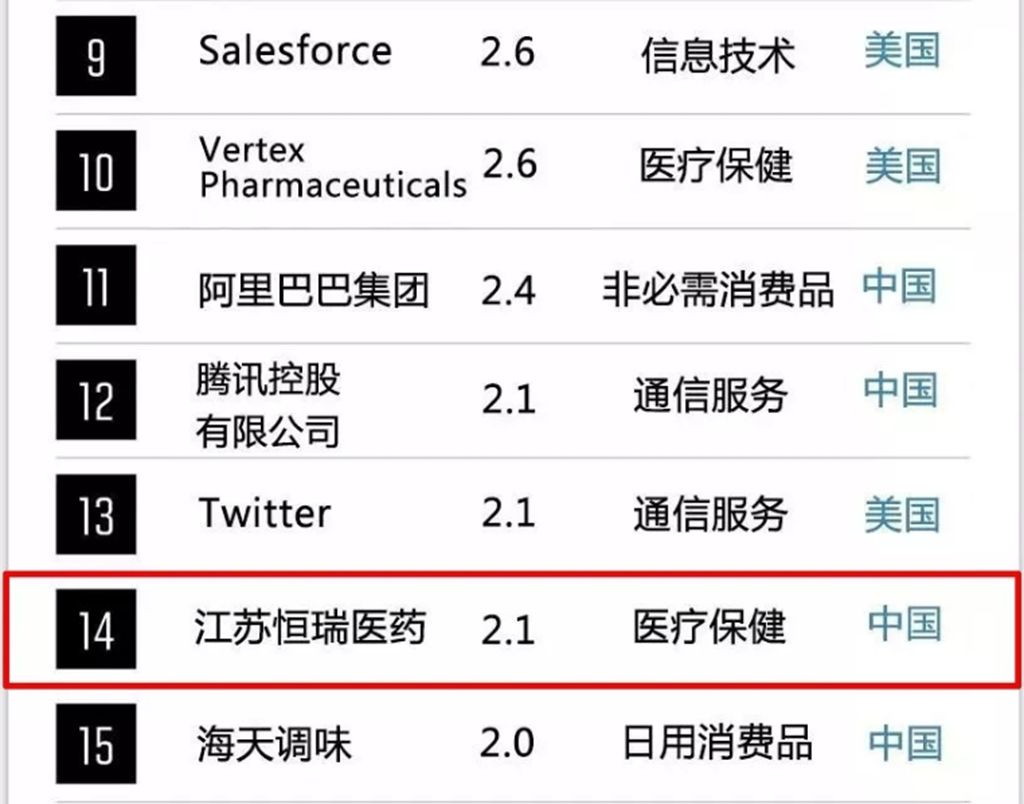
全球未来50强排行榜是《财富》杂志自2017年起与波士顿咨询公司(BCG)合作编制,以发现具有最大长期增长潜力的公司。今年通过对全球1000多家截止2018年底市值至少达到200亿美元的上市公司的市场潜力进行计算评估,同时从战略、技术和投资、人、架构等四个维度评估这些公司实现潜力的能力,利用广泛的金融和非金融数据源分析,形成这一长期前景最被看好的企业的排行榜。
恒瑞医药成立于1970年,全球员工20000多人,目前市值3600亿元,是国内市值最大的制药上市企业。多年来,公司坚持“科技创新”和“国际化”两大发展战略,立足生命科学和民生需求,努力为中国老百姓研制创新和高品质药品,提高药品可及性。公司每年投入销售额10%以上的资金用于研发,在美国、欧洲、日本和中国多地建有研发中心或分支机构,打造了一支3600多人的研发团队,其中海外员工300多人。
公司目前已有5个创新药艾瑞昔布、阿帕替尼、硫培非格司亭注射液、吡咯替尼和注射用卡瑞利珠单抗获批上市,1个创新药申报生产,并有多个创新药在美国开展临床。同时,恒瑞医药也是唯一一家将注射剂规模化销往欧美日市场的中国制药企业,现有包括注射剂、口服制剂和吸入性麻醉剂等在内的10多个产品获准在欧美日上市,造福全球患者。
海思科研发投入节节攀升,创新药即将迎来收获期
近年来,海思科研发投入节节攀升,年增长率逐年上升,研发投入占营业收入比重保持在10%以上。2019年上半年,公司研发投入2.29亿元,同比去年同期增长67.36%,研发投入占营业收入比重为12.23%。目前,在研制剂项目68个,其中仿制药58个,创新药7个,生物药2个,特殊医学用途配方食品1个。经历7年以上的布局和投入,海思科创新药即将迎来收获期,预计未来每年将有1-2个新药(含新适应症)获得批准。目前海思科共有7个1类新药在研,其中有3个属于神经系统疾病用药,麻醉用药HSK3486乳状注射液为丙泊酚me-better药物,目前已申请上市;用于带状疱疹后神经痛、糖尿病神经痛的HSK16149胶囊已进行到临床I期;术后急性止痛药HSK21542注射液已申报临床并正式获得受理。日前,海思科发布公告称,公司自主研发的1类新药HSK21542注射液的临床申请已正式获得受理,拟用于急慢性疼痛的治疗。该产品与公司早前提交上市的麻醉镇静1类新药HSK3486在商业推广上协同性强,且均具有良好的市场空间。据米内网数据,2018年中国公立医疗机构终端麻醉剂及止痛药合计销售额约304亿元。
国家医保局官宣:CHS-DRGs及其技术规范和分组方案
日前,国家医疗保障局发布了《关于印发疾病诊断相关分组(DRG)付费国家试点技术规范和分组方案的通知》(以下简称《通知》)。随《通知》一同发出的,还有《国家医疗保障疾病诊断相关分组(CHS-DRG)分组方案(核心组 ADRG)》(以下简称《分组方案》)与《国家医疗保障疾病诊断相关分组(CHS-DRG)分组与付费技术规范》(以下简称《技术规范》)。
同步开展DRG付费国家试点监测评估工作
在《通知》中,国家医保局要求,各试点城市要严格按照《技术规范》开展DRG相关工作,并在严格执行《分组方案》下,确保26个主要诊断分类(MDC)和376个核心DRG分组(ADRG)全国一致,按照统一的分组操作指南,结合各地实际情况,制定本地的细分DRG分组(DRGs)。各试点城市要坚持统分结合,不得随意更改MDC和ADRG组别。
其次,《通知》还对编码工作做了进一步的规定,要求各试点城市统一使用医保疾病诊断和手术操作、医疗服务项目、药品、医用耗材、医保结算清单等5项信息业务编码标准;推进信息业务编码标准落地应用;通过医保结算清单采集医疗机构有关数据;提高数据管理能力。
国家医疗保障局在北京成立DRG付费国家试点工作组技术指导组(以下简称“技术指导组”),并设在北京市医疗保障局。技术指导组将组织DRG付费国家试点专家组对省级、试点城市医保部门的骨干人员和核心专家进行培训。
医保局将同步开展DRG付费国家试点监测评估工作,重点是监测各试点城市工作进展,评估DRG付费改革成效,并采取适当方式公布有关结果。
同时,医保局也追本溯源,详细说明了为什么要制定CHS-DRG分组方案与CHS-DRG分组与付费技术规范。
世界药闻 TOP 3

全球首个埃博拉疫苗将在欧盟获批
10月21日,默克公司已收到欧洲药品管理局(EMA)的人类用医药产品委员会(CHMP)关于其V920埃博拉扎伊尔疫苗(rVSVΔG-ZEBOV-GP,减毒活疫苗)的积极意见,推荐有条件批准V920扎伊尔埃博拉疫苗。预计欧盟委员会(EC)将在第四季度作出审查决定。如果获得批准,这款疫苗的品牌名将为ERVEBO?,用于18岁及以上人群的主动免疫,以预防扎伊尔型埃博拉病毒(EbolaZaire)引起的埃博拉病毒病(EVD)。之前,EMA已授予V920优先药物资格(PRIME)。目前,V920也正在接受美国FDA的审查。该机构于今年9月中旬受理了V920的生物制品许可申请(BLA)并授予了优先审查,预计将在2020年3月14日作出审查决定。2016年,FDA还授予了V920突破性药物资格。除了与FDA和EMA密切合作,默沙东还向世界卫生组织(WHO)提交了获得资格预审的材料,并向非洲疫苗监管论坛(AVAREF)提交了材料。V920最初由加拿大公共卫生署(PHAC)开发,2010年将该产品授权给NewLink Genetics公司。默克公司于2014年(非洲西部埃博拉疫情达到高峰时)从NewLink Genetics获得了V920的许可,此后结成了各种联盟来开发该疫苗。V920采用了一种有缺陷的、能够感染家畜的水泡性口炎病毒,将病毒的一种基因用埃博拉病毒的基因替换。默沙东通过与一些外部合作者密切合作,在美国政府的部分资助下,开展了一项广泛的临床开发项目,其中包括美国卫生和人类服务部的生物医学高级研究及发展管理局(BARDA)以及美国国防部威胁减少管理局(DTRA)和联合疫苗采办计划(JVAP)等。V920的试验性供给活动由BARDA根据合同号HHSO100201700012C提供部分联邦资金支持。默沙东负责V920的研究、开发、制造及监管工作,该公司已承诺与其他利益相关方密切合作,加速疫苗的持续开发、生产和分销。
Stelara在美获批中重度活动性溃疡性(UC)新适应症
10月22日,强生(JNJ)旗下杨森制药宣布,美国FDA已批准抗炎药Stelara(ustekinumab),用于中度至重度活动性溃疡性结肠炎(UC)成人患者的治疗。值得一提的是,此次批准也是Stelara自2009年9月上市以来在美国方面的第5次批准。在欧盟,UC适应症于今年9月初获得批准。此次批准基于关键性III期UNIFI临床试验的数据,该试验达到了临床缓解的主要终点,结果显示,与安慰剂相比,Stelara诱导和维持临床缓解的患者比例显著提高。
Stelara于2009年9月获批上市,在美国监管方面已获批5个适应症:治疗中度至重度斑块型银屑病成人患者、治疗中重度斑块型银屑病(≥12岁)青少年患者、治疗活动性银屑病关节炎成人患者、治疗中重度克罗恩病(CD)成人患者、治疗中重度UC成人患者。
Opdivo和Yervoy +化疗在一线治疗NSCLC中达到终点
10月23日,Bristol-Myers Squibb(BMS)宣布,其免疫疗法Opdivo(nivolumab)和Yervoy(ipilimumab)联合2个疗程的化疗在一项三期CheckMate-9LA临床研究中的结果。该试验正在研究将免疫疗法联合化学疗法作为晚期非小细胞肺癌(NSCLC)的一线治疗,无论程序性细胞死亡配体1(PD-L1)表达如何。在预先专门的中期分析中,与最多进行四个化疗周期后再进行可选的维持治疗相比,Opdivo与Yervoy+化疗达到了其总生存期(OS)的主要终点。
药物治疗组的患者接受了长达两年的治疗,直到疾病进展或出现不可接受的毒性,而对照组的参与者接受了多达四个化疗周期的治疗。免疫疗法和化学疗法组合的安全性与其各自的已知特征相符。
研究的次要终点是无进展生存期(PFS)、总体应答率(ORR)和其他疗效指标。CheckMate-9LA试验的全部结果将在以后的会议上介绍。
BMS在多项试验中发现Opdivo联合化疗和Opdivo联合Yervoy无法满足一线NSCLC治疗的必要终点。但是,在将Opdivo与Yervoy和化学疗法相结合的III期CheckMate-227阶段的子分析中,达到了OS主要终点,这表明三联组合作为NSCLC患者的一线治疗方法可能是有效的。
(部分内容根据网络内容改编,知识产权归该权利人所有,若引用不当或版权争议发生,请相关方及时通知,我方将立即给予删除。)
整理:田田 学海无涯 编辑:绰绰



